Neuroendocrine transdifferentiation in breast cancer
Multi-omics integration identifying a master regulator and a monitorable, targetable therapy-escape state in HER2-positive breast cancer.
Co-first author (equal contribution) · under review at Cancer Discovery.
Problem
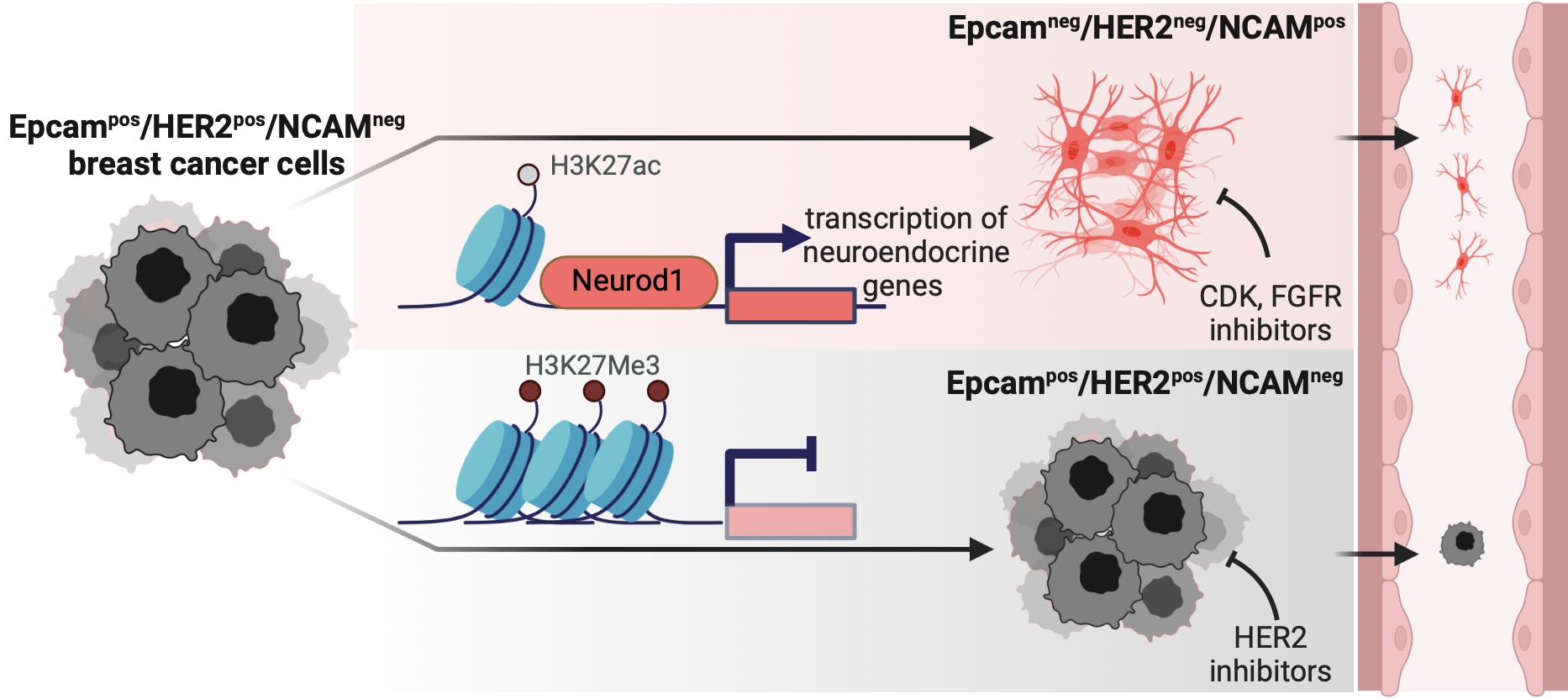
HER2-positive breast cancers can escape targeted therapy by transdifferentiating into an aggressive neuroendocrine state that no longer depends on HER2 — a lineage-plasticity program (epithelial-to-neuroendocrine transition) found in 22% of progressive metastatic cases in a 55-patient cohort. The questions: what molecular program drives the switch, can it be detected in patients, and is it therapeutically vulnerable?
Approach
I independently ran the complete computational analysis, integrating five orthogonal omics layers — whole-genome sequencing, RNA-seq, ATAC-seq, DNA methylation, and proteomics — across patient-derived organoid models sorted into epithelial and neuroendocrine sublines. The aim was to find the regulatory driver, not just a correlated marker: reconciling chromatin accessibility and histone marks with transcriptional and proteomic output, then validating causally rather than by correlation alone.
Result
The integration identified NEUROD1 as the master regulator of the escape state — pinpointed by motif enrichment and MYC ChIP-seq integration, then confirmed by CRISPR knockout and overexpression. HER2 turned out to be silenced epigenetically despite retained ERBB2 amplification, explaining the escape. We derived a 78-gene neuroendocrine signature (validated pan-cancer in lung and prostate datasets), nominated NCAM/CD56 as a liquid-biopsy biomarker for these EpCAM-negative cells, and uncovered an NCAM–FGFR1 dependency sensitising them to FGFR and CDK4/6 inhibitors where HER2 inhibitors fail. The work is under review at Cancer Discovery as a co-first-author paper.